Популярный рассказ о структуре и функции белка. О том, что мы знаем и о том, чего не знаем, а так же об имеющихся в этой области вычислительных задачах, требующих решения и интересных IT-специалистам. Сжато и тезисно, минимум воды.
Приятно видеть, что хабравчане регулярно интересуется другими предметными областями – например, биологией (более конкретно – структурой и функцией биологических макромолекул). Однако некоторые посты (например, этот ), вызывают у специалиста просто физическую боль из-за обилия совершенно диких фактологических ошибок. В этом посте мне хочется рассказать о структуре и функции белка. О том, что мы знаем и о том, чего не знаем, а так же об имеющихся в этой области вычислительных задачах, требующих решения и интересных IT-специалистам. Постараюсь рассказывать сжато и тезисно, чтобы информации было больше, а воды – меньше.
С химической точки зрения белок – это линейный (неветвящийся) полимер, состоящий из монотонно повторяющихся одинаковых блоков «основной цепи», к которым приделаны различные «боковые группы». Так как блоки основной цепи несимметричны, вся полипептидная цепь белка имеет направление, различают N- и C-конец полипептидной цепи.
Длина цепи – от 70 до более чем 1000 мономеров (аминокислотных остатков), средняя длина для высших организмов – примерно 500–600 аминокислотных остатков, для бактерий эта величина будет меньше, скорее 300–400 остатков. Всего в природе существует 20 стандартных аминокислот, одинаковых и для бактерии и для человека, то есть из основной цепи могут торчать 20 разных боковых групп.
(Тут возможна поправка – некоторые химические группы могут быть модифицированны после синтеза белка, например, фосфорилированы. Однако это не рассматривается как другая аминокислота, а рассматривается как продукт модификации исходной. Так же у высших организмов возможно встраивание двух неканонических аминокислот, но это редкое событие. То есть, строго говоря, разных аминокислот 22, из них 20 основных и 2 редкие, плюс некоторые боковые группы могут быть изредка химически модифицированы).
Из поколения в поколение генетическая информация передается в виде ДНК, в ней есть так называемые «белок-кодирующие области». В этих местах ДНК однозначным образом (для ботанов – с точностью до альтернативного сплайсинга и редактирования РНК) закодирована информация о линейной последовательности аминокислот для синтеза данного белка, плюс в клетке есть соответствующие машины, способные синтезировать белок по информации, изначально закодированной в ДНК.
Так как белок – линейный полимер, собранный из 20 стандартных мономеров, его так называемую «первичную структуру» легко представить в виде строки, например так:
>small ubiquitin-related modifier 3 precursor [Homo sapiens]
MSEEKPKEGVKTENDHINLKVAGQDGSVVQFKIKRHTPLSKLMKAYCERQG
LSMRQIRFRFDGQPINETDTPAQLEMEDEDTIDVFQQQTGGVPESSLAGHSF
Это аминокислотная последовательность маленького человеческого белка в формате FASTA, первая строчка, начинающаяся с «>», описывает его название, после чего следует последовательность аминокислот в соответствии со стандартной кодировкой (например, М –метиони, S – серин и тд, всего 20 букв стандартного однобуквенного кода), слева – N-конец белка, справа – его С-конец. Для разных белков длина строки будет очевидно разной, так как белки имеют разную длину. Последовательности всех известных белков можно найти в открытом доступе здесь: www.ncbi.nlm.nih.gov
3. Структура белка
Хорошо, с первичной структурой разобрались, но разве белок работает в развернутом линейном виде? Конечно нет. Тут надо заметить, что со структурной точки зрения есть разные классы белков: глобулярные, мембранные и фибриллярные. Мембранные белки, как следует из названия, живут только в клеточных мембранах, для стабилизации их структуры нужно особое окружение мембраны, мы не будем их рассматривать в этом обзоре. Фибриллярные белки имеют простое регулярное строение, похожи на вытянутые волокна, они не растворимы в воде и выполняют структурные функции (например, из кератина состоят волосы, к фибриллярным белкам относится белок из натурального шёлка). Недавно стали выделять класс разупорядоченных белков – белков, не обладающих постоянной трехмерной структурой, либо приобретающих ее только на короткое время при взаимодействии с другими белками. Наиболее интересный с практической точки зрения класс белков, который мы и будем рассматривать – глобулярные водорастворимые белки, к этому классу относится большинство белков.
Линейная полипептидная цепь в воде способна самопроизвольно сворачиваться в сложную трехмерную структуру (глобулу) и только в таком свернутом виде белки могут выполнять химический катализ и прочую интересную работу. Поэтому нам принципиально важно знать именно трехмерную укладку белка, так как только на этом уровне становится понятно, как белок работает.
Вопрос: сколько трехмерных структур соответствует конкретному белку?
Ответ: Одна, с точностью до небольшой подвижности маленьких «разупорядоченных» петель. Известно ровно одно исключение, когда одной последовательности соответствуют 2 достаточно разные структуры, это прионы .
Вопрос: Почему у белка только одна трехмерная структура?
*Ответ:** для химического катализа нам нужно расположить соответствующие химические группы строго определенным образом в пространстве. Для этого нужна жесткая структура. То есть весь белок должен быть жестким, чтобы поддерживать химические группы аминокислот активного центра в нужных местах (в реальности многие белки состоят из двух и более жестких частей, которые могут двигаться друг относительно друга, это нужно для регуляции активности белка (аллостерическая регуляция ), чтобы некий сигнал мог включать и выключать химическую активность белка-фермента). Чтобы структура была жесткой и стабильной, природа позаботилась о том, чтобы структура каждого белка соответствовала энергетическому минимуму данной системы атомов и этот минимум был настолько глубоким, чтобы белок из него не «выпрыгнул». Все другие, паразитные структуры, обладают большей энергией и белок все равно сваливается в энергетический минимум, соответствующий нативной структуре.
Вопрос: на чем держится трехмерная структура белка?
Ответ: если коротко, то в основном на большом количестве нековалентных взаимодействий. В принципе, химические группы белка могут образовывать: (1) водородную связь, эти группы есть и в основной цепи и у некоторых боковых групп, (2) ионную связь – электростатическое взаимодействие между разноименно заряженными боковыми группами, (3) Ван-дер-Ваальсово взаимодействие и (4) гидрофобный эффект, на котором держится общая структура белка. Суть в том, что в белке всегда есть гидрофобные ароматические остатки, им энергетически невыгодно контактировать с полярными молекулами воды, а выгодно «слипнуться» друг с другом. Таким образом, при сворачивании белка гидрофобные группы выталкиваются из водного окружения, «слипаясь» друг с другом и формируя «гидрофобное ядро», а полярные и заряженные группы, наоборот, стремятся в водное окружение, формируя поверхность белковой глобулы. Так же (5) боковые группы двух остатков цистеина могут образовать между собой дисульфидный мостик – полноценную ковалентную связь, жестко фиксирующую белок.
Соответственно, все аминокислоты делятся на гидрофобные, полярные (гидрофильные), положительно и отрицательно заряженные. Плюс цистеины, способные образовывать ковалентную связь между собой. Особыми свойствами обладают глицин – у него отсутствует боковая группа, сильно ограничивающая конформационную подвижность других остатков, поэтому он может очень сильно «гнуться» и находится в местах, где белковую цепь надо развернуть. У пролина же, наоборот, боковая группа образует кольцо, ковалентно связанное с основной цепью, жестко фиксируя ее конформацию. Пролины встречаются там, где надо сделать белковую цепь жесткой и негнущейся. Многие заболевания связаны с мутацией пролина на глицин, из-за чего структура белка слегка «плывет».
Вопрос: откуда вообще мы знаем о трехмерных структурах белка?
Ответ: из эксперимента, это абсолютно надежные данные.
Сейчас есть 3 метода для экспериментального определения структуры белка: ядерно-магнитный резонанс (ЯМР), cryo-EM (электронная микроскопия) и рентгеноструктурный анализ кристаллов белка.
ЯМР позволяет определить структуру белка в растворе, но он работает только для очень маленьких белков (для больших невозможно сделать деконволюцию).
Этот метод был важен для общего доказательства того, что у белка только одна трехмерная структура и что структура белка в кристалле идентична структуре в растворе. Это очень дорогой метод, так как требуется получить белок с изотопными метками.
Cryo-EM заключается в простой заморозке раствора белка и микроскопии. Минус метода – низкое разрешение (видна лишь общая форма молекулы, но не видно, как она устроена внутри), плюс плотность белка близка к плотности воды/растворителя, поэтому сигнал тонет в высоком уровне шума. В этом методе активно применяются компьютерные технологии работы с картинками и статистика для вытягивания сигнала из шума.
Отбираются миллионы картинок молекул белка, проводится разделение на классы в зависимости от ориентации молекулы относительно подложки, усреднение по классам, генерация eigenimages, новый раунд усреднения и так пока не сойдется. Потом из информации из разных классов можно восстановить трехмерный вид молекулы с низким разрешением. Если же есть внутренняя симметрия частиц (например, при cryo-EM анализе вирусов), то можно еще каждую частицу поусреднять в соответствии с операторами симметрии – тогда разрешение будет еще лучше, но хуже, чем в случае рентгеноструктурного анализа.
Рентгеноструктурный анализ – основной способ определения структур белка. Главный плюс – потенциально можно получить кристаллы даже очень больших комплексов из многих десятков белков (например, именно так была определена структура рибосомы – Нобелевская премия 2009 года). Минус метода – вначале нужно получить кристалл белка, но далеко не каждый белок хочет кристаллизоваться.
Зато после того, как кристалл получен, по дифракции рентгеновского излучения можно однозначно определить положения всех (упорядоченных) атомов в молекуле белка, этот метод дает самое высокое разрешение и позволяет в лучших случаях видеть позиции отдельных атомов. Было доказано, что структура белка в кристалле однозначно соответствует структуре в растворе.
Сейчас действует конвенция – если ты определил структуру белка любым из экспериментальных физических методов, структура должна быть помещена в открытый доступ в банк данных белковых структур (Protein Data Bank – PDB, www.pdb.org ), в настоящее время там находится более 90 000 структур (впрочем, многие из них повторяющиеся, например, комплексы одного и того же белка с разными малыми молекулами, такими, как лекарственные средства). В PDB все структуры лежат в стандартном формате, называющемся, внезапно, pdb. Это текстовый формат, в котором каждому атому структуры соответствует одна строчка, в которой указан номер атома в структуре, название атома (углерод, азот и тд), название аминокислоты, в которую входит атом, название цепи белка (A, B, C и тд, если это кристалл комплекса из нескольких белков), номер аминокислоты в цепи и трехмерные координаты атома в ангстремах относительно ориджина, плюс так называемые температурный фактор и заселённость (это сугубо кристаллографические параметры).
ATOM 1 N HIS A 17 –12.690 8.753 5.446 1.00 29.32 N
ATOM 2 CA HIS A 17 –11.570 8.953 6.350 1.00 21.61 C
ATOM 3 C HIS A 17 –10.274 8.970 5.544 1.00 22.01 C
ATOM 4 O HIS A 17 –10.193 8.315 4.491 1.00 29.95 O
ATOM 5 CB HIS A 17 –11.462 7.820 7.380 1.00 23.64 C
ATOM 6 CG HIS A 17 –12.551 7.811 8.421 1.00 21.18 C
ATOM 7 ND1 HIS A 17 –13.731 7.137 8.194 1.00 28.94 N
ATOM 8 CD2 HIS A 17 –12.634 8.384 9.644 1.00 21.69 C
ATOM 9 CE1 HIS A 17 –14.492 7.301 9.267 1.00 27.01 C
ATOM 10 NE2 HIS A 17 –13.869 8.058 10.168 1.00 22.66 N
ATOM 11 N ILE A 18 –9.269 9.660 6.089 1.00 19.45 N
ATOM 12 CA ILE A 18 –7.910 9.377 5.605 1.00 18.67 C
ATOM 13 C ILE A 18 –7.122 8.759 6.749 1.00 16.24 C
ATOM 14 O ILE A 18 –7.425 8.919 7.929 1.00 18.80 O
ATOM 15 CB ILE A 18 –7.228 10.640 5.088 1.00 20.22 C
ATOM 16 CG1 ILE A 18 –7.062 11.686 6.183 1.00 18.52 C
ATOM 17 CG2 ILE A 18 –7.981 11.176 3.889 1.00 24.61 C
ATOM 18 CD1 ILE A 18 –6.161 12.824 5.749 1.00 28.21 C
ATOM 19 N ASN A 19 –6.121 8.023 6.349 1.00 15.46 N
ATOM 20 CA ASN A 19 –5.239 7.306 7.243 1.00 14.34 C
ATOM 21 C ASN A 19 –4.012 8.178 7.507 1.00 14.83 C
ATOM 22 O ASN A 19 –3.431 8.715 6.575 1.00 18.03 O
ATOM 23 CB ASN A 19 –4.825 6.003 6.573 1.00 17.71 C
ATOM 24 CG ASN A 19 –6.062 5.099 6.413 1.00 21.26 C
ATOM 25 OD1 ASN A 19 –6.606 4.651 7.400 1.00 26.18 O
ATOM 26 ND2 ASN A 19 –6.320 4.899 5.151 1.00 31.73 N
Далее есть специальные программы, которые по данным из этого текстового файла могут графически отображать красивую трехмерную структуру молекулы белка, которую можно покрутить на экране монитора и, как говорил Гай Додсон, «дотронуться мышкой до молекулы» (например, PyMol , CCP4mg , старый RasMol ). То есть смотреть на структуры белка просто – ставишь программу, загружаешь нужную структуру из PDB и наслаждаешься красотой природы.
4. Анализируем структуру
Итак, мы поняли основную идею: белок — линейный полимер, сворачивающийся в водном растворе под действием множества слабых взаимодействий в стабильную и единственную для данного белка трехмерную структуру, и способный в таком виде выполнять свою функцию. Различают несколько уровней организации белковых структур. Выше мы уже познакомились с первичной структурой – линейной последовательностью аминокислот, которую можно выписать в строчку.

Вторичная структура белка определяется взаимодействием атомов основной цепи белка. Как уже было сказано выше, в состав основной цепи белка входят доноры и акцепторы водородной связи, таким образом, основная цепь может приобретать некоторую структуру. Точнее, несколько разных структур (детали все-таки зависят от различающихся боковых групп), так как возможно образование разных альтернативных водородных связей между группами основной цепи. Структуры бывают такие: альфа-спираль, бета-листы (состоящие из нескольких бета-тяжей), которые бывают параллельными и анти-параллельными, бета-поворот. Плюс часть цепи может и не иметь выраженной структуры, например в районе поворота петли белка. Эти типы структур имеют свои устоявшиеся схематичные обозначения – альфа-спираль в виде спирали или цилиндра, бета-тяжи в виде широких стрелок. Вторичную структуру удается достаточно достоверно предсказывать по первичной (стандартом является JPred ), альфа-спирали предсказываются наиболее точно, с бета-тяжами бывают накладки.
Третичная структура белка определяется взаимодействием боковых групп аминокислотных остатков, это и есть трехмерная структура белка. Можно представить себе, что вторичная структура сформирована и теперь эти спирали и бета-тяжи хотят уложиться все вместе в компактную трехмерную структуру, чтобы все гидрофобные боковые группы спокойно «слиплись» вместе в глубине белковой глобулы, сформировав гидрофобное ядро, а полярные и заряженные остатки торчали наружу в воду, формируя поверхность белка и стабилизируя контакты между элементами вторичной структуры. Третичную структуру изображают схематически несколькими способами. Если просто отрисовать все атомы, то получится каша (хотя когда мы анализируем активный центр белка, то мы хотим смотреть как раз на все атомы активных остатков).

Если мы хотим посмотреть, как устроен весь белок в общем, можно отобразить только некоторые атомы основной цепи, чтобы увидеть ее ход. Как вариант, можно нарисовать красивую схему, где поверх реального расположения атомов схематично нарисованы элементы вторичной структуры – так с первого взгляда видна укладка белка. После изучения всей структуры в общем, схематичном виде, можно отобразить химические группы активного центра и уже сосредоточиться на них. Задача предсказания третичной структуры белка – нетривиальная и в общем случае не решается, хотя может быть решена в частных случаях. Подробнее – ниже.
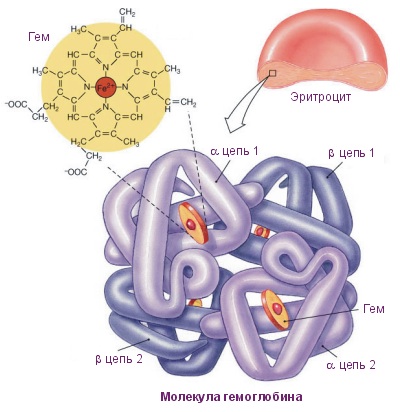
Четвертичная структура белка – да, есть и такая, правда не у всех белков. Многие белки работают сами по себе (мономеры, в данном случае под мономером имеется в виду одиночная свернутая полипептидная цепь, то есть белок целиком), тогда их четвертичная структура равна третичной. Однако достаточно много белков работает только в комплексе, состоящем из нескольких полипептидных цепей (субъединиц или мономеров — димеры, тримеры, тетрамеры, мультимеры), тогда вот такая сборка из нескольких отдельных цепей и называется четвертичной структурой. Самый банальный пример – состоящий из 4 субъединиц гемоглобин , самый красивый на мой взгляд пример – состоящий из 11 одинаковых субъединиц бактериальный белок TRAP .
5. Вычислительные задачи
Белок – сложная система из тысяч атомов, поэтому без использования компьютеров в структуре белка не разобраться. Задач, как решенных на приемлемом уровне, так и совсем не решенных, множество. Перечислю наиболее актуальные:
На уровне первичной структуры – поиск белков с похожей аминокислотной последовательностью, построение по ним эволюционных деревьев и тд – классические задачи биоинформатики. Главным хабом является NCBI — The National Center for Biotechnology Information, www.ncbi.nlm.nih.gov . Для поиска белков со сходной последовательностью стандартно используется BLAST: blast.ncbi.nlm.nih.gov/Blast.cgi
Предсказание растворимости белка. Речь идет о том, что если мы прочитаем геном какого-нибудь животного, определим по нему последовательности белков, переклонируем эти гены в кишечную палочку или baculovirus expression system, то окажется, что при экспрессии в этих системах примерно треть белков не будет сворачиваться в правильную структуру, и, как следствие, будет нерастворима. Тут выясняется, что большие белки на самом деле состоят из отдельных «доменов», каждый из которых представляет автономную, функциональную часть белка (несущую одну из его функций) и часто «вырезав» из гена отдельный домен, можно получить растворимый белок, определить его структуру и провести с ним опыты. Люди пытаются использовать машинное обучение (нейронные сети, SVM и прочие классификаторы), чтобы предсказывать растворимость белка, однако работает оно достаточно плохо (Гугл много чего покажет по запросу “protein solubility prediction” – есть много серверов, но по моему опыту все они работают отвратительно на моих белках). В идеале я хотел бы видеть сервис, который надежно сказал бы, где в белке находятся те самые растворимые домены, чтобы их можно было вырезать и работать с ними – такого сервиса нет.
На уровне вторичной структуры – предсказание той самой вторичной структуры по первичной (JPred )
На уровне третичной структуры – поиск белков со сходными трехмерными структурами (DALI , en.wikipedia.org/wiki/Structural_alignment ),
Поиск структур по заданной суб-структуре. Например, у меня есть расположение трех аминокислот активного центра в пространстве. Хочу найти структуры, которые содержать такие же три аминокислоты в таком же относительном расположении, либо найти структуры белков, мутирование которых даст возможность расположить нужные аминокислоты нужным образом. (гуглить «protein substructure search»)
Предсказание потенциальной подвижности трехмерной структуры, возможных конформационных изменений – normal mode analysis, ElNemo .
На уровне четвертичной структуры – предположим, известны структуры двух белков. Известно, что они образуют комплекс. Предсказать структуру комплекса (определить, как эти два белка будут взаимодействовать посредством shape matching, например). Гуглить «protein-protein docking»
6. Предсказание структуры белка
Выделил эту вычислительную задачу в отдельный раздел, ибо велика она, фундаментальна и не решается в общем случае.
Экспериментально мы знаем, что если взять белок, полностью развернуть его и бросить в воду, то он свернется обратно в исходное состояние за время от миллисекунд до секунд (это утверждение справедливо по крайней мере для небольших глобулярных белков без всяких патологий). Это значит, что вся информация, необходимая для определения трехмерной структуры белка, в неявном виде содержится в его первичной последовательности, поэтому так хочется научиться предсказывать трехмерную структуру белка по последовательности аминокислот in silico! Однако эта задача в общем случае не решена до сих пор. В чем же дело? Дело в том, что в первичной последовательности отсутствует в явном виде информация, необходимая для построения структуры. Во-первых, нет информации о конформации основной цепи – а она обладает значительной подвижностью, хотя и несколько ограниченной по стерическим причинам. Плюс каждая боковая цепь каждой аминокислоты может находиться в разных конформациях, для длинных боковых групп типа аргинина, это может быть больше десятка конформаций.
Что же делать? Есть достаточно известный хабравчанам самый общий подход, называемый «молекулярная динамика» и подходящий для любых молекул и систем. Берем развернутый белок, приписываем всем атомам случайные значения скоростей, считаем взаимодействия между атомами, повторяем до тех пор, пока система не придет в стабильное состояние, соответствующее свернутому белку. Почему это не работает? Потому что современные вычислительные мощности позволяют за месяцы работы кластера считать десятки наносекунд для системы из тысяч атомов, какой является белок, помещенный в воду. Время же сворачивания белка – миллисекунды и больше, то есть вычислительных мощностей не хватает, разрыв – в несколько порядков. Впрочем, пару лет назад американцы совершили некоторый прорыв. Они использовали специальное железо, оптимизированное для векторных вычислений и после оптимизации на аппаратном уровне у них за месяцы работы машины получилось посчитать молдинамику до миллисекунд для очень маленького белка и белок свернулся, структура соответствовала экспериментально определенной ( en.wikipedia.org/wiki/Anton_(computer)) )! Однако праздновать победу еще рано. Они взяли очень маленький (его размер раз в 5–10 меньше среднего белка) и один из самых быстросворачивающихся белков, классический модельный белок, на котором изучалось сворачивание. Для больших белков время расчетов увеличивается нелинейно и потребуются уже годы, то есть еще есть над чем работать.
Другой подход реализован в Rosetta . Они разбивают последовательность белка на очень короткие (3–9 остатков) фрагменты и смотрят, какие конформации для этих фрагментов присутствуют в PDB, после чего запускают Монте-Карло по всем вариантам и смотрят, что получится. Иногда получается что-то годное, но в моих случаях через несколько дней работы кластера получаешь такой бублик, что возникает немой вопрос: «Кто писал их оценочную функцию, ставящую какую-то хорошую оценку вот этой загогулине?».
Есть инструменты и для моделирования вручную – можно предсказать вторичную структуру и попробовать вручную крутить ее, находя лучшую укладку. Некие гениальные люди даже выпустили игрушку FoldIt , представляющую белок схематично и позволяющую укладывать его, как-бы собирая головоломку (для интересующихся структурой – рекомендую!). Есть абсолютно официальное соревнование для предсказателей белковых структур, называемое CASP . Суть в том, что когда экспериментаторы определяют новую структуру белка, не имеющую аналогов в PDB, они могут не выкладывать ее сразу в PDB, а выставить последовательность этого белка на конкурс предсказаний CASP . Через некоторое время, когда все закончат свои предсказательные модели, экспериментаторы выкладывают свою экспериментально определенную структуру белка и смотрят, насколько хорошо сработали предсказатели. Самое интересное, что игроки FoldIt, не будучи учеными, как-то выиграли CASP у профессионалов моделирования белковых структур и предсказали структуру белка точнее. Однако даже эти успехи не позволяют утверждать, что проблема предсказания структуры белка решается – очень часто модель очень далека от реальной структуры.
Все это относилось к моделированию белков ab initio, когда нет никакой априорной информации о структуре. Однако очень часто бывают ситуации, когда для некоторого белка в PDB присутствует его отдаленный родственник с уже известной структурой. Под родственником подразумевается белок с похожей первичной последовательностью. Считается, что для белков со сходством по первичной последовательности больше 30% одинаковая укладка основной цепи (хотя одинаковая укладка наблюдалась и для белков, не проявляющих никакого статистически достоверного сходства по первичной последовательности). В случае наличия гомолога (похожего белка) с известной структурой, можно сделать «гомологичное моделирование», то есть попросту «натянуть» последовательность твоего белка на известную структуру гомолога, а потом погонять минимизацию энергии, чтобы как-то все это дело утрясти. Такое моделирование показывает хорошие результаты при наличие очень близких гомологов, чем дальше гомолог – тем больше ошибка. Инструменты для гомологичного моделирования – Modeller, SwissModel.
Можно решать и другие задачи, например, пытаться моделировать, что произойдет, если внести в белок ту или иную мутацию. Например, если заменить гидрофильную аминокислоту на поверхности белка на другую гидрофильную, то скорее всего структура белка не изменится вообще. Если заменить аминокислоту из гидрофобного ядра на другую гидрофобную, но другого размера, то скорее всего укладка белка останется той же, но слегка «съедет» на доли ангстрема. Если же заменить аминокислоту из гидрофобного ядра на заряженную, то скорее всего белок просто «взорвется» и не сможет свернуться.
Может показаться, что все не так уж и плохо и мы достаточно хорошо пониманием сворачивание белка. Да, мы понимаем кое-что, например до некоторой степени мы понимаем общие физические принципы, лежащие в основе сворачивания полипептидной цепи – они рассматриваются в замечательном учебнике Птицына и Финкельштейна «Физика белка». Однако это общее понимание не позволяет нам ответить на вопросы «Свернется ли данный белок или не свернется?», «Какая структура будет у этого белка?», «Как сделать белок с желаемой структурой?».
Вот одна из иллюстраций: мы хотим локализовать один из доменов большого белка, это стандартная задача. У нас есть фрагмент, который сворачивается и растворим, то есть это живой и здоровый белок. Мы же хотим найти его минимальную часть и начинаем методами генетической инженерии с обоих концов удалять по 2–3 аминокислоты, экспрессировать такой обрезанный белок в бактерии и смотреть его сворачиваемость экспериментально. Мы делаем десятки конструкций с такими маленькими делециями и видим такую картину – полностью растворимый и живой белок отличается от полностью мертвого и несворачивающегося на 3 аминокислоты. Повторюсь, это объективный экспериментальный результат. Проблема в том, что сейчас не существует вычислительного метода, который предсказал бы сворачиваемость белка хотя бы на уровне «да/нет» и сказал мне, где проходит граница между сворачивающимся и несворачивающимся белком, потому мы вынуждены клонировать и экспериментально проверять десятки вариантов. Это лишь одна из иллюстраций того, что наше понимание структуры белка весьма далеко от совершенства. Как говорил Ричард Фейнман, «Чего не могу воссоздать, того не понимаю».
Так что, господа программисты, физики и математики, нам еще есть над чем работать.
На этой оптимистичной ноте разрешите откланяться, благодарю всех, кто осилил сей опус.
Для глубоко знакомства с предметной областью рекомендую следующий минимум:
- «Физика белка» Птицын и Финкельштейн. Большую часть материала Алексей Витальевич Финкельштейн выложил в онлайн, чем и рекомендую с благодарностью воспользоваться: phys.protres.ru/lectures/protein_physics/index.html (а я утащил оттуда несколько картинок)
- Патрушев, «Искусственные генетические системы», особенно часть II «Белковая инженерия». Есть на торрентах в формате Djvu
- Для информации, опубликованной в биологических научных журналах, есть официальный поисковик PubMed ( www.pubmed.org ) — у него стоит попросить почитать про «protein engineering» и тому подобное.
http://habrahabr.ru/post/181850/
 Ученым из Школы медицины Перельмана Университета Пенсильвании (Perelman School of Medicine, University of Pennsylvania) и Медицинского центра Хадасса Еврейского университета (Hadassah-Hebrew University Medical Center) удалось ускорить размножение бета-клеток человеческой поджелудочной железы, вырабатывающих жизненно важный для организма гормон инсулин, пересадив донорские клетки в организм мышей. Но сначала они подавили в них активность одного из белков. Новые клетки сохраняют характерные черты зрелых бета-клеток и демонстрируют физиологическую реакцию на глюкозу. Свое исследование ученые опубликовали в журнале The Journal of Clinical Investigation.
Ученым из Школы медицины Перельмана Университета Пенсильвании (Perelman School of Medicine, University of Pennsylvania) и Медицинского центра Хадасса Еврейского университета (Hadassah-Hebrew University Medical Center) удалось ускорить размножение бета-клеток человеческой поджелудочной железы, вырабатывающих жизненно важный для организма гормон инсулин, пересадив донорские клетки в организм мышей. Но сначала они подавили в них активность одного из белков. Новые клетки сохраняют характерные черты зрелых бета-клеток и демонстрируют физиологическую реакцию на глюкозу. Свое исследование ученые опубликовали в журнале The Journal of Clinical Investigation. В бета-клетках с подавленным p57 ДНК может реплицироваться. Розовым показано ядро, прошедшее репликацию ДНК, зеленым – инсулин, синим – ядра,
В бета-клетках с подавленным p57 ДНК может реплицироваться. Розовым показано ядро, прошедшее репликацию ДНК, зеленым – инсулин, синим – ядра, Структуры белка амилина, полученные с помощью методов молекулярной динамики.
Структуры белка амилина, полученные с помощью методов молекулярной динамики.
 «Грязные» мышцы. За 1, 3 и 5 недель (слева направо) в мышечных волокнах контрольных мух (верхний ряд) накопились большее количество белковых
«Грязные» мышцы. За 1, 3 и 5 недель (слева направо) в мышечных волокнах контрольных мух (верхний ряд) накопились большее количество белковых






{kind=link}
{kind=link}